generalitet
Fabry sykdom er en sjelden arvet genetisk lidelse forårsaket av mutasjonen av GLA genet.

Figur: struktur av alfa-galaktosidase A.
GLA-genet er plassert på X-kromosomet og koder for et enzym kalt alfa-galaktosidase A. Dette enzymet har en grunnleggende rolle i prosessen med å bryte ned en lipid, kjent som globotriesosylceramid.
Hos mennesker med Fabrys sykdom virker enzymet alfa-galaktosidase A dårlig; tilsvarende. globotriesosylceramidmolekylene har en tendens til å akkumulere unormalt i noen intracellulære organeller - lysosomene - med alvorlig lidelse for de involverte cellene.
Fabry sykdom er ansvarlig for kliniske manifestasjoner ved nevrologisk, dermatologisk, okular, gastrointestinal, cerebrovaskulær, nyre- og kardial nivå.
For å diagnostisere Fabrys syndrom med absolutt sikkerhet er en hensiktsmessig genetisk test avgjørende.
For tiden er det ingen terapier som er i stand til å kurere Fabry-syndrom, men bare behandlinger av symptomatisk karakter (dvs. rettet mot å lindre symptomer).
Blant disse symptomatiske behandlingene, er det viktigste enzymreparasjonsbehandling, som innebærer administrasjon av en analog, opprettet i laboratoriet, av enzymet alfa-galaktosidase A.
Hva er Fabrys sykdom?
Fabry sykdom er den arvelige genetiske sykdommen som følge av akkumulering i blodvegger i vev og organer av en bestemt type lipid, kalt globotriesosilceramid .
Fabry sykdom er en sfingolipidose og er en del av den heterogene gruppen av såkalte lysosomale lagringssykdommer .
Betydningen av sfingolipidose og lysosomal lagringssykdom
Kort lysosomale lagringssykdommer er en gruppe på ca. 50 sjeldne arvelige sykdommer, som i fellesskap har funksjonsfeil hos en bestemt kategori av intracellulære organeller: lysosomer . Denne feilen avhenger av et enzymmangel og fører til unormal akkumulering, i selve lysosomene, av lipider eller glykoproteiner, med tilhørende tap av cellulær funksjon.
Ved å dreie seg til sfingolipidose er disse spesielle lysosomale lagringssykdommer, karakterisert ved den skadelige akkumulering, innenfor lysosomene, av noen spesifikke sfingolipider .
En sfingolipid er en lipid, hvis molekylære formasjon innebærer et sfingosinmolekyl .
Hva er oppgaven med lysosomer?
Lysosomer er små vesikler, som vanligvis finnes i eukaryotiske celler, som har til oppgave å nedbryte og fordøye fremmede molekyler, makromolekyler eller stoffer som ikke lenger er nyttige for cellen.
Lysosomer kan defineres som fordøyelsessystemet i den eukaryote cellen.
Andre navn på Fabry sykdom
Fabrys sykdom er kjent av flere andre navn, som er:
- Anderson-Fabry sykdom ;
- Diffus angiokeratoma ;
- Alfa-galaktosidase A-mangel .
årsaker
Forutsetning: Det menneskelige kromosomale settet inneholder to sexkromosomer: X-kromosomet og Y-kromosomet .
To spesielle kombinasjoner av X og Y bestemmer kjønn for et individ: XX- kombinasjonen, som karakteriserer det kvinnelige kjønet, og XY- kombinasjonen, som skiller den mannlige kjønn .
Fabry sykdom oppstår på grunn av mutasjoner i GLA genet, lokalisert på sex kromosom X.
Under normale forhold (derfor i fravær av mutasjoner) koder dette genet (dvs. produserer) et lysosomalt enzym, kalt alfa-galaktosidase A, som har en grunnleggende rolle i å bryte ned nevnte globotriesosylceramid, en sfingolipid, til enklere komponenter.
Hos mennesker med Fabrys sykdom produserer det muterte GLA-genet mindre alfa-galaktosidase A enn nødvendig, og dette innebærer en progressiv akkumulering av intakt (dvs. ikke nedbrutt) globotriesosylceramidmolekyler inne i lysosomene med tilhørende skade og lidelse for de involverte cellene.
Med andre ord, mens det hos mennesker med sunn GLA vi ser riktig dekomponering av globotriesosylceramid inne i lysosomene, er det i mennesker med mutert GLA den samme prosessen med dekomponering som ikke er tilstrekkelig til å møte behovene til en celle, han er syk.
Hvilke organer og vev påvirkes mest av GLA-mutasjoner
I emner med Fabrys sykdom er cellene i menneskekroppen som er mest påvirket av GLA-mutasjoner, :
- Cellene som utgjør veggen av blodkar,
- Nyre celler,
- Hjerteceller e
- Cellene i nervesystemet (nevroner).
Denne informasjonen er viktig for å forstå årsaken til symptomatologien som preger Fabrys sykdom.
arv
Fabry sykdom er et eksempel på en X-koblet recessiv arvelig sykdom, akkurat som hemofili .
Særligheten av recessive arvelige sykdommer knyttet til X-kromosomet er at for å kunne manifestere seg med alvorlige symptomer, må alle X-kromosomene i hver celle ha det samme muterte genet. Dette har flere implikasjoner:
- Kvinner med bare ett X-kromosom som presenterer den genetiske mutasjonen er generelt i perfekt helse (unntatt i sjeldne tilfeller), men bærere av den arvelige sykdommen.
I disse individene - som kalles "sunne bærere", er den totale eller nesten totale mangelen på en symptomatologi på grunn av det faktum at det sunne X-kromosomet kompenserer for den uregelmessige oppførselen til det muterte X-kromosomet.
- Mannlige personer som er født fra foreningen mellom en sunn mann og en sunn bærer har 50% sjanse for å være syk. Disse fagene arver X-kromosomet fra moren, og dette kromosomet kan enten være den muterte eller den sunne.
Hos menn finnes det beskrevne kromosomale kompensasjonsfenomenet ikke for sunne bærere, da X-kromosomet er bare en.
- De eneste syke kvinner er kvinner med begge muterte X-kromosomer. I disse situasjonene er det faktisk ingen kromosomal kompensasjon, og situasjonen er sammenlignbar med den for mannen med hans X-kromosom som presenterer den genetiske mutasjonen.
Det skal spesifiseres at tilstedeværelsen av to muterte X-kromosomer i en kvinne er en reell sjeldenhet, da det krever den usannsynlige foreningen mellom en syke mann og en sunn transportkvinne.
- I lys av hva som ble sagt i de forrige punktene om kromosomal kompensasjon og usannsynligheten for foreningen mellom en syke mann og en sunn bærer, er recessive arvelige sykdommer knyttet til X-kromosomet mye lettere observerbare hos menn.
Fabrys sykdom respekterer, nesten helt, hver av de ovennevnte punkter: Den eneste uoverensstemmelsen - hvis vi vil kalle det her - gjelder friske bærere, som hyppigere viser en ubetydelig symptomatologi i forhold til hva som skjer for andre arvelige sykdommer av samme skriver.
epidemiologi
Fabry sykdom er en av de vanligste lysosomale lagringssykdommene.
Hvis det er et relativt pålitelig estimat på grunn av forekomsten i den mannlige befolkningen (1 tilfelle for hver 40.000-60.000 nyfødte), på grunn av forekomsten i den kvinnelige befolkningen er det svært få pålitelige data.
Ifølge noen studier relatert til diffusjonen i de ulike løpene i verden, virker det som at Fabrys sykdom rammer alle verdens etniske grupper, med en liten preferanse for den hvite rase.
Symptomer og komplikasjoner
Fra et symptomatologisk synspunkt opptrer Fabrys sykdom tidlig i barndommen, med svært milde manifestasjoner som er vanskelige å fange, noen ganger til og med på et erfarent øye.
I løpet av årene blir problemene tydeligere og berørte mennesker lider mer; På dette stadiet er det enda enklere å identifisere sykdommen av leger.
De typiske symptomer og tegn på Fabrys sykdom er:
- Smerter i ekstremiteter av lemmer (hender og føtter) og gastrointestinale smerter .
Også kjent som akroparestesi, synes smerte i ekstremiteter av lemmer å være knyttet til skade på noen perifere nerver. Gastrointestinal smerte, derimot, synes å stamme fra en funksjonsfeil i blodkarene som leverer organene i mage-tarmkanalen;
- Mørke flekker på huden . Disse stedene vises i klynger og kalles angiokeratomer ;
- Anhidrose, som er manglende evne til å utsette svette, eller hypohydrosis, som er den unormale reduksjonen i svette;
- Korneal opacity . Også kjent som hornhinnen verticillata, korneal opacity som skiller Fabry sykdom ikke kompromittere pasientens visuelle evner;
- Høreproblemer . Blant disse er tinnitus og en viss grad av døvhet;
- Svimmelhet ;
- Kvalme ;
- Sans for tilbakevendende tretthet ;
- Diaré .

Figur: angiokeratom i en pasient med Fabry syndrom.
Komplikasjoner av Fabrys sykdom
På et avansert stadium har Fabry-sykdommen en tendens til å involvere viktige anatomiske organer og strukturer, som nyrer, hjerte og cerebrale blodkar, forårsaker dem skade og forringe deres funksjon.
De viktigste konsekvensene av involvering av nyrer, hjerte og cerebrale blodårer, av Fabry sykdom, er:
- Hjertesykdommer, for eksempel hjerteinfarkt, hjertearytmier, valvulopatier, restriktiv kardiomyopati, hjertesvikt etc. De er resultatet av den massive akkumuleringen av globotriesosilceramid i hjertecellene og fører ofte til døden.
- Nyresvikt . På samme måte som tilfelle av hjertesykdommer, kommer den fra den massive akkumuleringen av globotriesosylceramid i nyrene og representerer en viktig dødsårsak.
- Predisponering til episoder av trombose, hjerneslag, forbigående iskemiske angrep, etc.
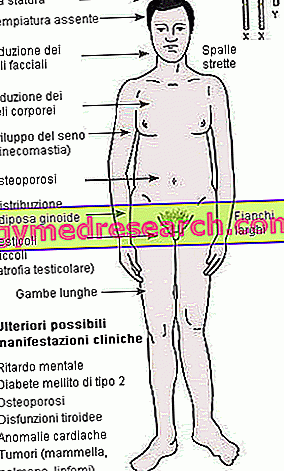
Komplett samling av alle symptomer, tegn og komplikasjoner av Fabrys sykdom | |
| Neurologiske manifestasjoner |
|
Dermatologiske manifestasjoner |
|
Gastrointestinale manifestasjoner |
|
Okulære manifestasjoner |
|
Cerebrovaskulære manifestasjoner |
|
Kardiale manifestasjoner |
|
Nyre manifestasjoner |
|
diagnose
Fabry sykdom er en vanskelig tilstand for å diagnostisere, spesielt i det første tiåret av pasientens liv.
Generelt, for å formulere en korrekt diagnose av Fabrys sykdom, er følgende grunnleggende: En grundig fysisk undersøkelse, en forsiktig historie, studien av pasientens familiehistorie, den enzymatiske analysen for å måle aktiviteten av alfa-galaktosidase A i leukocytter og en genetisk test.
Sistnevnte er den diagnostiske testen som eliminerer tvil, i tilfelle mistanke, og som gjør det mulig å fastslå nøyaktige konnotasjoner av mutasjonen mot GLA genet.
terapi
Behandlingen av Fabrys sykdom består av en substitusjonsenzymbehandling, som innebærer administrering til pasienten av en kopi opprettet i laboratoriet av det opprinnelige manglende enzymet, nemlig alfa-galaktosidase A.
Dermed er formålet med enzymutskiftningsterapi å gi pasienter med noe som er i stand til å etterligne funksjonene til det normale, fysiologiske enzymet.
Enzymutskiftingstest, studert for Fabrys sykdom, tillater ikke behandling av sistnevnte - Dessverre gjenstår GLA-mutasjonen gjennom livet - men tillater lindring av hele symptomatologien (spesielt smerte) med god resultater. For mer informasjon, se medisinarkene:
- Fabrazyme
- Replagal
- Galafold
Andre behandlinger
Det er svært vanlig at, foruten enzymutskiftningsterapi, foreskriver legene også:
- Administrasjon av smertestillende midler;
- Tar medisiner mot gastrointestinale problemer
- Administrasjon av blodfortynnende legemidler og legemidler for regulering av hjerterytmen;
- En farmakologisk behandling mot hypertensjon;
- Dialyse.
I nærvær av alvorlig nedsatt nyrefunksjon kan basene eksistere for å benytte seg av nyretransplantasjon .
prognose
For menn med Fabry syndrom er gjennomsnittlig levetid 58, 2 år, sammenlignet med 74, 7 år for friske menn.
For kvinner med Fabrys syndrom er gjennomsnittlig forventet levetid 75, 4 år sammenlignet med 80 år for friske kvinner.
Som det fremgår av disse tallene, hos kvinner, er sykdommen mindre alvorlig enn hos menn.
Blant personer med Fabrys syndrom er hjertesykdom den viktigste dødsårsaken.