generalitet
Noonan syndrom er en sjelden, noen ganger arvelig, genetisk lidelse som endrer den normale utviklingen av ulike anatomiske deler av kroppen. Uvanlige ansiktsegenskaper, kort statur og noen medfødte hjertefeil er de viktigste patologiske og diagnostiske tegnene.

Konsekvensene av patologien er ikke alltid dramatisk: i noen tilfeller kan Noonan syndromet forårsake en nyansert symptomatologi og tillate et normalt liv.
Kort påminnelse om genetikk: DNA og kromosomer
Før det beskrives Noonan syndrom, er det godt å lage en kort referanse til genetikk.
KROMOSOMER OG DNA
Hver celle av et sunt menneske har 23 par homologe kromosomer : 23 er mor, det er arvet fra moren, og 23 er faderlige eller arvet fra faren. Et par av disse kromosomene er seksuelt, det vil si at det avgjør individets kjønn; De resterende 22 parene, i stedet, består av autosomale kromosomer . I sin helhet inneholder de 46 menneskelige kromosomer hele genetisk materiale, bedre kjent som DNA . I et individs DNA skrives hans somatiske egenskaper, hans predisposisjoner, hans fysiske evner etc.
GENER OG MUTASJONER AV DNA
DNA er organisert i mange sekvenser, mer eller mindre lenge, kalt gener . Hvert gen har et spesifikt kromosom og dets motstykke, som det er til stede i to eksemplarer, kalt alleler . En allel kommer fra moren og ligger i mors kromosom; Den andre allelen kommer fra faren og er plassert i faderkromosomen.

Figur: Organisering av et gen i et par homologe kromosomer. Et par homologe kromosomer inneholder spesifikke gener, som alle har to varianter, allelene, opptar samme kromosomale posisjon og gjør de samme funksjonene (unntatt mutasjoner). Det venstre paret av kromosomer har to like alleler (begge blå); det rette paret har i stedet to forskjellige alleler (den ene er rød, den andre er blå).
Fra gener genererer proteinene, presentert i kroppen vår. Når en DNA-mutasjon oppstår, kan et gen (vanligvis en allel) av et gitt kromosom være defekt og derfor produsere et defekt protein.
Hva er Noonan syndrom
Noonan syndrom er en genetisk, noen ganger arvelig sykdom som forårsaker flere anatomiske anomalier (og ikke bare) i flere deler av kroppen. De viktigste endringene forekommer i ansikt, hjerte og skjelett, men det er mulig at reproduktive systemet, lymfesystemet, huden, nervesystemet, øynene, ørene, blodet og nyrer.
Unormalitetene som karakteriserer Noonan syndrom er ikke alltid det samme hos alle pasienter: i noen er de uskarpe, så mye at de går nesten ubemerket og gir et nesten normalt liv; i noen andre, på den annen side, er de så accentuert at de truer de som bærer dem.
epidemiologi
Noonan syndrom er uvanlig: det påvirker faktisk ett barn hvert 2500 nyfødte. Hanner og kvinner er like berørt og det er ikke et etnisitet spesielt mer predisponert enn andre.
årsaker
Noonan syndrom er en genetisk sykdom, derfor er det forårsaket av en endring i DNA . Forskerne er ganske sikre på at minst åtte gener er involvert, men de kan fortsatt ikke klart forklare hvordan de forårsaker anatomiske abnormiteter.
Hva gjør disse åtte genene?
ROLLE
Generene involvert i Noonan syndrom har en spesiell fysiologisk rolle: de produserer proteiner som tillater vekst og utvikling av våre celler. Med andre ord, disse proteinene er regulatoriske ender, som tar vare på livet og skjebnen til hver enkelt celle i detalj.
Når gener som lager disse proteinene muterer, endres også de cellulære reguleringsmekanismer som er beskrevet ovenfor, med alvorlig skade på kroppen.
GENENE INVOLVED
Av de åtte genene som er involvert i Noonans syndrom, har bare fire blitt fullt identifisert, da de endres med en statistisk merkbar frekvens.
De er:
- PTPN11 : dens mutasjon karakteriserer rundt 50% av tilfellene. Den ligger på kromosom 12, og oppdagelsen av dens implikasjon i Noonan syndrom går tilbake til 2001. Proteinet, som det produserer, har en grunnleggende rolle under embryonisk utvikling: det styrer faktisk veksten, differensiering og deling av celler, de spesielt i hjertet. Vi vet sikkert at en mutert allel er nok til at sykdommen skal vises (dominans).
- SOS1 : mutert i 10-15% av tilfellene. Det ble forstått at han kunne være involvert i sykdommen i 2006.
- RAF1 : mutert i 5-10% av tilfellene. Hans rolle var ikke kjent før 2007.
- KRAS : mutasjonen karakteriserer ca. 2% av tilfellene. Oppdagelsen av dens implikasjon er nylig: 2006.
Som du kan se, er forskningen relatert til disse genene ganske nylige, og dette forklarer hvorfor de fortsatt er ventende.
INHERITANCE?: JA ELLER IKKE?
Genetiske mutasjoner, i 50% tilfeller, er sporadiske (dvs. på grunn av tilfeldighet), mens de resterende 50% overføres av en av foreldrene .
Vanligvis er de foreldrene som overfører sykdommen til barna, ikke kjent med at de er rammet av Noonan syndrom, da dette ikke er en alvorlig og åpenbar form.
symptomer
Noonan syndrom manifesterer seg med flere karakteristiske tegn i flere deler av kroppen.
Disse tegnene består av mer eller mindre utprøvde anatomiske anomalier og i mer eller mindre alvorlige patologiske forhold. Faktisk manifesterer noen pasienter sykdommen klart, i alle henseender; andre, derimot, er mer nyanserte eller begrensede til enkelte nettsteder.
HOVED OG SEKUNDARISKE PATOLOGISKE EGENSKAPER
Uvanlige ansiktsegenskaper, kort statur og en rekke medfødte hjertefeil er de abnormiteter som finnes i alle de som lider av Noonan syndrom; Av denne grunn vurderes sykdommens hovedkarakteristika (NB: det medfødte begrepet betyr til stede fra fødselen ).
I stedet betraktes følgende anomalier som sekundære egenskaper, da de er mindre vanlige enn de forrige:
- Lett utseende av hematomer og blødninger
- Læringsproblemer og adferdsmessige uregelmessigheter
- Visuelle mangler av ulike slag
- lymfødem
- Muskelhypotoni
- Hørselstap
- Infertilitet og unormal kjønnsapparat
- Vanskelig fôring (i barndommen)
- Skjelett anomalier
BEHANDLINGENE AV FACE
De unormale ansiktsfunksjonene kan bli lagt merke til allerede når pasienten er svært liten; Over tid blir deres variasjon observert, og blir permanent i voksen alder.
- Svært tidlig barndom (mindre enn et år): Øynene er fjernt fra hverandre og har en tendens mot bunnen. Ørene er lave og orientert mot baksiden av hodet. Sporet, som er tilstede over overleppen, er dypt; nakken er kort og håret bak hodet er lavt.
- Barndom : øynene begynner å bli fremtredende, mens øyelokkene senkes ( ptosis ) og blir tykkere. Nesen knuses ved roten, men har en bred base og en bulbespiss.
- Barndom : De forrige karakteristikkene legges til en inexpressivitet av ansiktet, forstørrede lepper og en større lengde av ansiktet.
- Ungdom : pannen blir bred og haken peker og gjør ansiktet til en trekant. Noen ansiktsegenskaper er fremhevet, for eksempel linjene som starter fra nesen og når munnens hjørner, mens øynene blir mindre fremtredende. Den korte halsen er beriket med andre detaljer: de første foldene ( pterigium colli ) vises og trapezius muskler er mer voluminøse.
- Voksne alder : furene som går fra nesen til munnens sider er dype og tydelige. Munnen blir enda mer fremtredende og huden blir rynket og klar. Øynene er alltid fjerne og med øyelokkene tykkere og påvirket av ptosis.
Lav status
Vekt og lengde ved fødselen er normal. Imidlertid er det i de første 18-24 månedene observert vekst og forsinkelse i forhold til barn i samme alder.

Figur: Ansiktet til en voksen med Noonan syndrom. Fra nettstedet: login.aafp.org
Dette skyldes et hormonelt problem ( veksthormon, GH ), men også til noen vanskeligheter, som barnet har i å spise.
Vekstretardering observeres også i barndommen og fremfor alt under puberteten, når gutter har en tendens til å utvikle seg plutselig. Forskjeller fra jevnaldrende på denne tiden i livet er åpenbare.
I fravær av behandling når høyden hos menn i gjennomsnitt 162 cm, mens det på kvinner er 153 cm. Med de rette terapeutiske behandlinger kan høyden også falle innenfor normal gjennomsnitt.
KONGENITALCARDIAC DEFEKTER
80% av pasientene med Noonan syndrom er født med mer eller mindre alvorlige hjertefeil. Disse anomaliene kan være:
- Stenose i lungeventilen : Dette er en innsnevring (stenose) av hjerteventilen (som gjør at blodet kan strømme fra hjerteets høyre ventrikel mot lungearterien). Lungearterien er ansvarlig for å transportere blod til lungene på grunn av oksygenering.
- Stenose i lungearterien : det er en innsnevring av lungearterien; Denne feilen, som den forrige, begrenser mengden blod som når lungene for oksygenering.
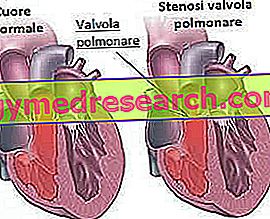
- Feil i inter ventrikulær septum (mellom ventrikkene) eller inter-atrielle (mellom atriene): består i dannelse av et hull mellom vævvegget som separerer ventriklene (eller det som separerer atriene). Dette fører til at blodet strømmer mellom de to rommene, forårsaker sirkulasjonsproblemer, noen ganger til og med alvorlige.

ANDRE ANOMALIER
Da dette er mange hendelser, vil vi prøve å beskrive dem i hovedtrekkene.
- Læringsproblemer og adferdsmessige uregelmessigheter . Intelligens kan være lavere enn gjennomsnittet. Imidlertid er det mange individer som, til tross for at de er bærere av sykdommen, har en normal IQ.
Det samme gjelder for atferd: I noen tilfeller er pasienten irritabel, gjentar andre menneskers lyder og ord og har merkelige kostholdskrav.
- Lett utseende av hematomer og blødninger . Hos 50% av pasientene reduseres koagulasjonsevnen. Derfor er blodtap, selv etter trivielt traumer, iøynefallende. Det er en egenskap som ikke skal overses når man tar visse stoffer (aspirin) eller gjennomgår operasjon (alt fra den mest invasive til enkel utvinning av en tann).
- Visuelle mangler av ulike slag . Omtrent halvparten av pasientene har synproblemer av ulike slag. Strabismus, astigmatisme, nærsynthet, lat øye ( amblyopi ), hypermetropi og nystagmus er mulige visuelle defekter som kan oppstå hos en pasient med Noonan syndrom.
- Lymphedem . Dette er en defekt i lymfesystemet . Lymfevæske, eller lymf, akkumuleres i enkelte områder av kroppen, for eksempel hender og føtter. Det skjer hovedsakelig i barndommen.
- Muskelhypotoni . Dette er en reduksjon i muskeltonen. Det er typisk for barndommen og barndommen.
- Hørselstap . Dette er en midlertidig episode under ungdommen. Det skyldes hyppig otitis i mellomøret.
- Infertilitet og unormal kjønnsapparat . Genital lidelser observeres hos 60% av pasientene. Ofte er individene menn, som lider av kryptorchidisme (dvs. sviktet av en eller begge testikler ned i pungen). For å løse dette problemet, må kirurgi brukes. Ellers, det er uten inngrep, har individet redusert antall spermatozoer, derfor er han mindre fruktbar.
Kvinner med Noonan syndrom har vanligvis ikke disse problemene.
- Vanskelig fôring . Det er et typisk barndomsproblem og bidrar til å forsinke veksten. Barnet har problemer med brystmelkssuging og har en tendens til å kaste opp etter hvert måltid.
- Skjelett anomalier . Spesielt hos unge ungdommer, kan artikulær hypermobilitet (dvs. ledd med et vidt spekter av bevegelser), skoliose, bryst excavatum (eller trakt), brystet bli sett og brystvorter på avstand på en uvanlig måte.
HVOR OG HVORFOR SKAL jeg henvise til en lege?
Hvis du merker ansiktsfunksjoner som de som er beskrevet ovenfor i et barn, er det tilrådelig å få den lille pasienten til å gjennomgå en pediatrisk undersøkelse. Legen vil da foreta de riktige vurderingene og, hvis han mistenker Noonan syndrom, vil utføre de nødvendige diagnostiske testene.
I noen tilfeller er de anatomiske anomaliene uskarpe, slik at de som er bærere av sykdommen ikke vet at de er berørt. Men selv i disse situasjonene er diagnosen viktig, på grunn av hjerteproblemer som kan oppstå.
komplikasjoner
Komplikasjoner relatert til Noonan syndrom avhenger av graden av alvorlighetsgrad av sykdommen. Derfor utgjør hver pasient ut fra dette synspunkt en sak i seg selv.
Blant de viktigste komplikasjonene fortjener de et sitat:
- Alvorlig intellektuell forsinkelse
- Ekstremt nedsatt blodkoagulasjonskapasitet
- Lymphedem dannelse rundt vitale organer, som for eksempel hjertet og lungene
- Alvorlige anatomiske anomalier i kjønnsapparatet, med repercussjoner på urinen (spesielt nyrene)
diagnose
En sykdom som Noonan syndrom, karakterisert ved et stort antall spesielle tegn, kan diagnostiseres selv etter en objektiv undersøkelse . Men i noen tilfeller er sykdommen ikke helt tydelig, slik at det, som det allerede er sagt ved flere anledninger, kan gå ubemerket til puberteten eller voksenalderen (når pasientens første seksuelle behov oppstår).
I alle fall, for å klargjøre tvil, er det svært pålitelige genetiske tester, som den såkalte karyotypen .
Når sykdommen er diagnostisert, er det viktig å overvåke de patologiske forholdene som er potensielt farlige for pasienten, for eksempel hjertefeil eller reduserte koagulasjonsevner ved spesifikke tester. Avhengig av utfallet av disse testene kan alvorlighetsgraden av hvert tilfelle vurderes nøyaktig.
De viktigste tegnene i den fysiske undersøkelsen:
| Overvåking utføres av:
|
behandling
Å være en genetisk sykdom, er Noonan syndrom ikke herdbart.
Imidlertid kan symptomatologien som den produserer begrenses og lindres gjennom ganske effektive terapier.
HELSE AV HJØRPROBLEMER
Å vite hvilke lidelser som plager hjertet, er viktig for å kunne sette den mest hensiktsmessige behandlingen.
Enkelte hjertemessige anomalier kan kontrolleres med en enkel farmakologisk behandling (diuretika, antiarytmika og beta-blokkere); Andre alvorligere (for eksempel stenosen i lungeventilen eller defektene i septumet) kan kreve kirurgi.
I alle fall er periodisk overvåking avgjørende, da en tilsynelatende uskadelig situasjon kan forvandle seg til noe veldig alvorlig og dramatisk utvikling.
HORMONAL CARE FOR VEKST
Ofte er redusert statural utvikling på grunn av dårlig veksthormonproduksjon, eller GH . Derfor har eksogen administrering av en av dets analoger ( somatropin ), skapt i laboratoriet, gode resultater, forutsatt at det terapeutiske programmet følges nøye. Sistnevnte er veldig enkelt: daglige injeksjoner, fra 3-5 år i livet.
Bivirkninger er sjeldne og består hovedsakelig av kløe og rødhet i injeksjonsområdet.
Periodisk overvåkning av hormonnivåer anbefales.
VEDLEGG AV Læringsdefekten
I noen tilfeller kan Noonan syndrom betydelig forringe intellektuelle fakulteter. I slike tilfeller trenger pasienten gyldig støtte, spesielt i skolen.
ANDRE BEHANDLINGER
For visuelle defekter kan briller som er egnet for den diagnostiserte patologien være tilstrekkelig; det er sjelden å måtte ty til kirurgi.
Når det gjelder koagulasjonsproblemer, finnes det stoffer som fremmer koagulasjon, som skal administreres i behovstid. Videre er en anbefaling å huske ikke å ta antikoagulanter, for eksempel aspirin og dets derivater.
Hvis lymphedem er dannet rundt kritiske organer som hjerte eller lunger, kan lymfevæske dreneres ved å sette inn et spesialrør. Kirurgi, i disse tilfellene, er en sjelden mulighet.
Endelig, med hensyn til infertilitet og kjønnsdefekter, er det mulig å løse criptorchidisme med en spesifikk intervensjon (NB: leseren er påminnet om at infertilitet i Noonans syndrom er et typisk mannlig problem).
prognose
Prognosen varierer fra pasient til pasient. I noen individer forårsaker Noonan syndromet en nyansert symptomatologi og tillater et nesten normalt liv (forutsatt at passende behandlinger blir brukt); i andre, kompromitterer det imidlertid alvorlig intellektuell kapasitet og generell helse.
Derfor kan prognosen være positiv (første sak), men også negativ (andre tilfelle).
Ved siden av disse to ekstreme situasjonene blir moderate tilfeller satt inn, det vil si midt mellom de ikke-seriøse skyggeformene og de alvorlige. På grunn av disse forholdene, for at prognosen skal være positiv, er tidlig diagnose viktig etterfulgt av tilstrekkelig og tidlig behandling. Først da kan prognosen ha en positiv innvirkning.
FOREBYGGING
Dessverre er det ikke mulig å forhindre sporadiske mutasjoner. Arvelige former kan derimot forebygges ved å informere friske personer, som lider av Noonan syndrom, om muligheten for å overføre sykdommen til barna sine.


