generalitet
Craniosynostose er begrepet som leger indikerer en abnormitet av skallen på grunn av for tidlig fusjon av en eller flere kraniale suturer.
Cranial suturer er de fibrøse leddene som knytter sammen knivene til kranialhvelvet (dvs. de frontale, tidsmessige, parietale og oksipitale beinene).
Craniosynostose kan være et isolert fenomen (ikke-syndromisk kraniosynostose) eller resultatet av noen spesielle sykelige tilstander (syndromisk kraniosynostose). Blant de sykelige tilstandene som forårsaker en for tidlig fusjon av kraniale suturer, er de mest kjente: Crouzons syndrom og Apert syndrom.
Med den for tidlige fusjonen av kraniale suturer, har hjernestrukturene ikke den rette plassen til å vokse. Dette har ulike konsekvenser, inkludert hovedsakelig økningen i intrakranielt trykk (intrakranial hypertensjon).
En rask og nøyaktig diagnose gjør det mulig å planlegge en ad hoc- behandling. Sistnevnte er av kirurgisk type og har som endelige målsetting separasjonen av fusesi suturer tidlig.
Minner om anatomien til menneskeskallen
Utstyrt med bein og brusk, skallen er skjelettstrukturen av hodet som utgjør ansiktet og beskytter hjernen, hjernen, hjernestammen og sensoriske organer.

For å forenkle studien og forståelsen av skallen har anatomistene tenkt å dele den i to rom, kalt neurokranium og splanknokranium .
neurocranium
Neurokraniet er den øvre kraniale regionen, som inneholder hjernen og noen av de viktigste sensoriske organene. Dens viktigste bein - strengt flatt - er frontal, temporal, parietal og occipital bein; Disse, tatt sammen, danner den såkalte kranialhvelvet .
splanchnocranium
Den splankokranium, eller ansiktsmasse, er den antero-underordnede delen av skallen, sammensatt av jevne og ujevne ben. Representerer skjelettstrukturen i ansiktet, derfor inneholder den benete elementer som mandal, overkjeve, kinnben, neseben osv.
Hva er craniosynostose?
Craniosynostose er en sjelden anomali av skallen, preget av en unaturlig form av hodet på grunn av for tidlig fusjon av en eller flere kraniale suturer . Cranial suturer er de fibrøse leddene som knytter sammen knivene til kranialhvelvet (dvs. de frontale, tidsmessige, parietale og oksipitale beinene).

Fra nettstedet: //www.wkomsi.com/
NÅR SKAL LUKTEN AV KRANALSUTURER FØRE?
Under normale forhold skjer fusjonen av kranial sutur i postnatal perioden (NB: Noen prosesser endrer seg selv ved 20 års alder). Denne lange fusjonsprosessen gjør at hjernen kan vokse og utvikle seg riktig.
Hvis fusjonen, som i tilfelle av kraniosynostose, foregår for tidlig - derfor under prenatal, perinatal * eller tidlig barndom - gjennomgår de encephale elementer (hjerne, cerebellum og hjernestamme) og noen følelsesorganer (spesielt øyne) en forandring av form og vekst.
årsaker
Den patofysiologiske prosessen som bestemmer kraniosynostose er den for tidlige fusjonen av kraniale suturer .
Denne prosessen kan representere et isolert fenomen - hvor med "isolert" menes det at det ikke er forbundet med en bestemt sykelig tilstand - eller det kan være konsekvensen av enkelte syndromer, nesten alltid av genetisk natur.
I lys av dette har leger besluttet å klassifisere kraniosynostose i to kategorier:
- Ikke-syndromisk kraniosynostose . Uttrykket ikke-syndromisk indikerer at kranial anomali ikke er forbundet med noen patologi eller annen fysisk defekt.
- Syndromisk kraniosynostose . Begrepet syndrom betyr at kranial misdannelse er resultatet av et bestemt syndrom, i de fleste tilfeller av en genetisk type.
NON-SINDROMIC CRANIOSINOSTOSIS
Leger og forskere har ennå ikke oppdaget hva årsakene til ikke-syndromisk kraniosynostose er.
De foreslo ulike hypoteser - inkludert påvirkning av miljøfaktorer eller hormonelle typer problemer - men ingen av disse teoriene fant bekreftelse i eksperimentelle resultater.
Derfor, for å forstå den presise opprinnelsen til avviket, er det behov for videre studier i denne forbindelse.
CRANIOSINOSTOSI SINDROMICA
Ifølge den siste medisinske forskningen er det mer enn 150 forskjellige syndromer, alle ganske sjeldne, som kan forårsake craniosynostose.
Blant disse syndromene er de mest kjente og mest vanlige:
- Crouzons syndrom . Resultatet av spesifikke mutasjoner i generne FGFR2 (kromosom 10) og FGFR3 (kromosom 4), påvirker denne spesielle morbid tilstanden en nyfødt hvert 60.000 og fører til tilstedeværelsen av anomalier utelukkende på nivået av hodet og ansiktet.
- Apert syndrom . Det oppstår hovedsakelig på grunn av mutasjoner i FGFR2-genet (det samme som i Crouzons syndrom) og påvirker en nyfødt hver 100.000.
I motsetning til Crouzons syndrom er de genetiske endringene av FGFR2 slik at misdannelser ikke bare omfatter skallen og ansiktet, men også hender og føtter.
- Pfeiffer syndrom . Det oppstår på grunn av mutasjoner i det "vanlige" FGFR2-genet og et gen med lignende funksjoner, kalt FGFR1 (kromosom 8). Egenheten ved disse mutasjonene er at - i tillegg til deformiteter av skallen og ansiktet - de også bestemmer: syndaktisk, brachydactyly og tommelfinger og store tær (uforholdsmessig til de andre fingrene).
Pfeiffer syndrom har en forekomst av ett tilfelle per 100.000 nyfødte.
- Saethre-Chotzen's syndrom . Det er en genetisk tilstand som påvirker et nyfødt hver 50.000 eller så. Forårsaker ulike misdannelser i skallen, ansiktet, hender og føtter. Noen spesifikke mutasjoner av TWIST1-genet, lokalisert på kromosom 7, er ansvarlige for oppstarten av Saethre-Chotzen-syndromet.
EPIDEMIOLOGI AV CRANIOSINOSTOSI
På grunnlag av den nyeste statistikken virker det som om et barn lider av craniosynostose hver 1800-3000 eller så.
Med hensyn til det mest berørte kjønn har flere kliniske studier vist at 3 av 4 pasienter er menn. Årsaken til at craniosynostose er mer utbredt i den mannlige befolkningen er helt ukjent.
Craniosynostose risikofaktorer.
- Lav fødselsvekt
- For tidlig fødsel
- Avansert paternal alder
- Mor røyking under graviditet
Symptomer og komplikasjoner
De fleste symptomene som observeres i nærvær av kraniosynostose skyldes en økning i trykk i skallen . I medisin kalles økt trykk i skallen intrakranial hypertensjon eller intrakranial hypertensjon .
I nærvær av kraniosynostose er intrakranial hypertensjon en følge av at hjernen og andre strukturer inne i skallen ikke har riktig plass til å vokse, så de går for å presse på hodehårene i hodet.
Når det er sagt, er det viktig å huske at hvis de involverte kraniale suturene er mange eller om tilstanden ikke behandles i tide, kan craniosynostose føre til redusert utvikling av kognitive fakulteter og lav IQ.
Symptomer på endokransk hypertensjon
De mulige symptomene på intrakranial hypertensjon er:
- Vedvarende hodepine. Vanligvis blir det verre om morgenen og om natten.
- Visjonsproblemer. De består av dobbeltsyn, sløret syn og sløret syn.
- oppkast
- irritabilitet
- Puffy eller fremtredende øyne
- Vanskelighetsgrad ved å følge bevegelsen av objekter
- Høreproblemer
- Åndedrettsproblemer
- Endringer i mental status
- papilledema
Antall kraniale suturer involvert i utviklingen av kraniosynostose har en avgjørende innflytelse på forekomsten av intrakranial hypertensjon.
For eksempel har leger observert at involvering av en enkelt kranial sutur induserer intrakranial hypertensjon hos 15% av pasientene; mens involvering av minst to suturer fører til en økning i trykk i skallen hos minst 60% av pasientene.
I nærvær av en mild form for kraniosynostose begynner intrakraniell hypertensjon å være problematisk, forårsaker den nevnte symptomatologien, rundt 4-8 år av livet.
Tegn på CRANIOSINOSTOSI
Blant tegnene til craniosynostose er de vanligste:- Formasjoner av stive rygger langs kranial suturene
- Abnormaliteter av kraniale fontaneller
- Hodet med dimensjoner som ikke står i forhold til resten av kroppen
Typer CRANIOSINOSTOSIS
Hodeformen til pasienter med kraniosynostose avhenger av hvilke kraniale suturer som har stengt for tidlig.
Etter å ha observert dette, anså legene det hensiktsmessig å skille mellom craniosynostose i ulike typer, avhengig av de involverte kraniale suturer.
Typer av craniosynostose er:
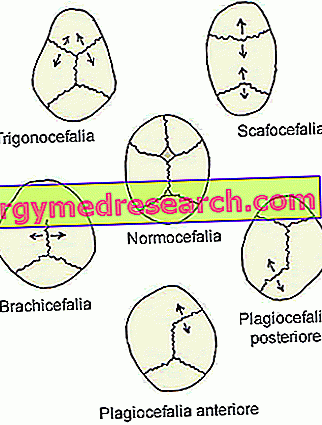
- Sagittal synostose ( dolichocephaly eller scaphocephaly ). Det er den vanligste typen av craniosynostose; Faktisk karakteriserer den omtrent halvparten av de kliniske tilfellene.
Dens tilstedeværelse sammenfaller med for tidlig lukning av sagittale kraniale suturer, plassert i den øvre delen av skallen, mellom parietalbeinene.
Fra //en.wikipedia.org/wiki/Plagiocephaly
- Coronal craniosynostosis ( brachycephaly ) Det er den nest vanligste typen av craniosynostose; karakteriserer om ett klinisk tilfelle hver fjerde.
Utbruddet involverer den for tidlige sammensmeltingen av koronale suturene, som løper mellom frontalbeinet og parietalbeinene.
- Metopisk synostose ( trigonocephaly ). Det er en svært sjelden type craniosynostose, som bare skiller 4-10% av tilfellene.
Utseendet sitt faller sammen med den for tidlige fusjonen av metopisk (eller frontal) sutur, som går fra nesen til den øvre delen av hodet, og separerer frontbenet i to. Generelt siver denne suturen naturlig i det sjette livet i livet.
- Den lambdoid-synostose ( plagiocephaly ). Det er den sjeldneste typen craniosynostose. Faktisk er det bare 2-4% av kliniske tilfeller.
Dens tilstedeværelse innebærer tidlig sammensmelting av lambdoid sutur, som befinner seg mellom parietalbeinene og oksepitalbenet, på baksiden av hodet.

KOMPLIKASJONER
I tillegg til å forringe intellektuell utvikling, kan en ubehandlet kraniosynostose bestemme:
- Det såkalte obstruktiv søvnapné-syndromet .
- Permanente ansiktsendringer, spesielt i øyne og ører.
- Permanente deformiteter ved bunnen av skallen (for eksempel misdannelsen, eller Arnold-Chiari syndromet).
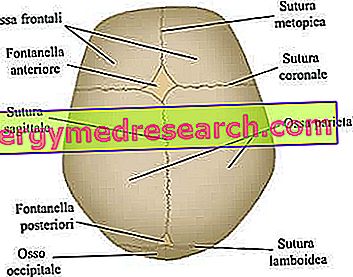
De viktigste kraniale suturer involvert i craniosynostose prosessen. Fra nettstedet: www.sciencebasedmedicine.org
- Hydrocephalus .

diagnose
For å diagnostisere craniosynostose, fysisk undersøkelse, evaluering av klinisk historie og radiologiske bilder levert av røntgenbilder eller CT-skanning til hodet er avgjørende.
Hvis craniosynostosen er av syndromisk type, er det også viktig å etablere den sykelige tilstanden som førte til utbruddet. Derfor kan legene ty til blodprøver og fremfor alt genetisk rådgivning .
EKSAMINERINGSMÅL
Den fysiske undersøkelsen består av en nøye analyse av legen av de kliniske tegnene som er tilstede på motivets hode, mistenkt for å lide av kraniosynostose.
Generelt er en barnelege ansvarlig for å gjennomføre denne viktige diagnostiske kontrollen.
KLINISK HISTORIE
Evalueringen av klinisk historie er viktig for diagnostiske formål, da det inneholder spørsmål relatert til risikofaktorene for kraniosynostose.
Derfor vil legen (vanligvis alltid en barnelege) undersøke om:
- Barnet er født for tidlig eller med lav vekt.
- Hva var farenes alder på tidspunktet for unnfangelsen.
- Hvis moren røykt under graviditeten.
RADIOLOGISKE TEST
Røntgenstråler og CT i hodet tjener mer enn noe for å bekrefte diagnosen og vise legen hvilke kraniale suturer har smeltet for tidlig.
Kunnskapen om hvilke kraniale suturer som er involvert, gjør det mulig å planlegge den mest hensiktsmessige kirurgiske behandlingen.
behandling
Craniosynostose er herdbar bare ved kirurgisk inngrep .
Sistnevnte består av en operasjon av separasjon av kraniale suturer som fusjonerer forgjengelig fra hverandre.
Det endelige terapeutiske målet med kirurgi er å gi de encefaliske strukturer og noen fornuftige organer, for eksempel øynene, med den plassen som trengs for å utvikle og fungere som sitt beste.
BESTE TID TIL INTERVENT
Det er ingen total avtale, fra leger, om hva som er den beste tiden å utføre craniosynostosis kirurgi.
Ifølge noen eksperter vil den ideelle perioden for operasjonen være i sen barndom, når risikoen for en gjentakelse er lavere (dvs. en annen for tidlig fusjon av kranialsuturene). I tilfelle av tilbakefall, må operasjonen gjentas, og dette anbefales ikke, gitt delikatessen av prosedyren.
Ifølge andre eksperter vil den mest hensiktsmessige tiden være tidlig i barndommen (mellom 6 og 12 måneder av livet), når skallen ennå ikke er fullstendig forenet og beinene fortsatt er formbare. Muligheten for å forme ben (smidighet) gjør det mulig å løse morfologiske abnormiteter i beinene, noe som kan forårsake alvorlige estetiske feil og funksjonelle problemer (i kjeve eller øyne) i en mer moden alder.
Mulige kirurgiske tilnærminger
Det er to forskjellige kirurgiske tilnærminger: tradisjonell kirurgi, også kalt "åpen" og endoskopisk kirurgi.
- Tradisjonell (eller "åpen") kirurgi .
Det forutsetter generell anestesi (derfor pasienten er ubevisst under hele operasjonen) og utøvelsen av et kirurgisk snitt på hodet, akkurat på det punktet hvor de radiologiske bildene viste kranial anomali.
Gjennom snittet på hodet fjerner operasjonen kirurg (en nevrokirurg) den unormale bein og overlater den til en spesialist i kraniofacial kirurgi, som modifiserer den og gir den en form som muliggjør normal utvikling av hjernestrukturene.
Etter endringen setter neurosurgen på benet i sin opprinnelige posisjon og lukker snittet med masker.
Som mange tradisjonelle operasjoner er den "åpne" operasjonen noe invasiv; For å gjøre det fordelaktig er det imidlertid å kunne modifisere beinstrukturen nøyaktig og med gode resultater.
- Endoskopisk kirurgi inngrep .
Det innebærer bruk av et endoskop, et verktøy som ligner på et fleksibelt rør, utstyrt med et fiberoptisk kamera i den ene enden og koblet til en skjerm.
Fra operasjonelt synspunkt består det i å sette endoskopet inn i en åpning som er gjort på skallen og i separasjonen, med selve endoskopet, av fusesi suturen forutgående.
Nevrokirurgen klarer å orientere seg inne i hodet, takket være bildene som kameraet prosjekterer på den eksternt tilkoblede skjermen.
Endoskopisk kirurgi inngripen er avgjort mindre invasiv enn den "åpne" operasjonen (sykehusinnleggingsperioden er også kortere), men den har to ulemper: den er kun indikert for pasienter i noen få måneder (6 generelt), den som har formbare bein; har større risiko for tilbakefall.
POST-OPERATIV FASE
Vanligvis må en pasient med kraniosynostose, som blir utsatt for kirurgi, forbli på sykehuset i ca 4-5 dager etter operasjonen. I løpet av denne tiden overvåker nevrokirurgen og hans medarbeidere periodisk deres vitale tegn og kontrollerer at alt går bra.
Etter oppsigelsen er det planlagt en rekke periodiske kontroller, som først, halvårlig og deretter med veksten av pasienten årlig.
prognose
Prognosen avhenger av ulike faktorer, blant annet:
- Årsakene som induserte craniosynostose. Noen genetiske sykdommer som er ansvarlige for denne anomali er svært alvorlige og har en dårlig prognose.
- Stillingen av suturene fusjonerte tidlig. Hvis suturene ligger i stillinger som, for nevrokirurgen, er "ubehagelige" for å nå, blir kraniosynostoseintervensjonen komplisert og kan ikke gi de ønskede resultatene.


