generalitet
Prader-Willi syndrom er en sjelden genetisk lidelse som forårsaker fysiske, atferdsmessige og intellektuelle abnormiteter. De mest karakteristiske kliniske tegnene er fedme (og de relaterte sykdommene) og redusert muskelton.
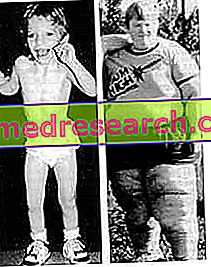
Fysisk undersøkelse er vanligvis tilstrekkelig til å etablere riktig diagnose, men pålitelige genetiske tester kan også utføres.
Dessverre er det fortsatt ingen endelig terapi; Noen farmakologiske og adferdsmessige tiltak kan imidlertid begrense den tilknyttede symptomatologien.
Kort påminnelse om genetikk
Før det beskrives Prader-Willi syndrom, er det godt å lage en kort referanse til genetikk.
KROMOSOMER OG DNA
Hver celle av et sunt menneske har 23 par homologe kromosomer : 23 er mor, det er arvet fra moren, og 23 er faderlige eller arvet fra faren. Et par av disse kromosomene er seksuelt, det vil si at det avgjør individets kjønn; De resterende 22 parene, i stedet, består av autosomale kromosomer . I sin helhet inneholder de 46 menneskelige kromosomer hele genetisk materiale, bedre kjent som DNA . I et individs DNA skrives hans somatiske egenskaper, hans predisposisjoner, hans fysiske evner etc.
GENER OG MUTASJONER AV DNA
DNA er organisert i mange sekvenser, mer eller mindre lenge, kalt gener .

Figur: Organisering av et gen i et par homologe kromosomer. Et par homologe kromosomer inneholder spesifikke gener, som alle har to varianter, allelene, opptar samme kromosomale posisjon og gjør de samme funksjonene (unntatt mutasjoner). Det venstre paret av kromosomer har to like alleler (begge blå); det rette paret har i stedet to forskjellige alleler (den ene er rød, den andre er blå).
Hvert gen har et spesifikt kromosom og dets motstykke, som det er til stede i to eksemplarer, kalt alleler . En allel kommer fra moren og ligger i mors kromosom; Den andre allelen kommer fra faren og er plassert i faderkromosomen.
Fra gener genererer proteinene, presentert i kroppen vår. Når en DNA-mutasjon oppstår, kan et gen (vanligvis en allel) av et gitt kromosom enten være defekt og derfor produsere et defekt protein eller mangler helt.
Hva er Prader-Willi syndrom?
Prader-Willi syndrom er en sjelden genetisk lidelse preget av en rekke fysiske, intellektuelle og atferdsdefekter avhengig av en forandring av kromosom 15.
Under tidlig barndom manifesterer seg syndrom med uvanlig muskel svakhet og utviklingsforsinkelse. Senere i barndommen begynner andre problemer å oppstå, for eksempel kontinuerlig appetitt, læringsproblemer og atferdsavvik.
Personer med Prader-Willi syndrom er, svært ofte, mennesker som lider av fedme, hvorav ulike hjerteproblemer oppstår. Dette er hovedårsaken til døden.
epidemiologi
Prader-Willi syndrom er en sjelden sykdom: Faktisk er født et berørt barn registrert i gjennomsnitt hver 15.000-30.000 nyfødte.
Det påvirker menn og kvinner lik og har ingen forkjærlighet for visse raser.
årsaker
Årsaken til Prader-Willi syndromet er en genetisk mutasjon på kromosom 15 . Det nøyaktige genet som er slått, er ennå ikke blitt avklart; Mistanke faller, mer enn noe annet, på en kromosomal region, som inkluderer flere gener.

Figur: Et kromosom 15 og generene mistenkt for å ha en rolle i Prader-Willi syndrom. Fra nettstedet: www.kreatech.com
GENENE PÅ CHROMOSOMA 15
Vanligvis bruker kroppens celler både begge alleler til å lage proteiner. Med andre ord betyr dette at både kromosomer, mor og far er nyttige og gir deres genetiske bidrag.
Imidlertid, i noen spesielle celler, på grunn av et evolusjonært og ikke-patologisk problem, virker bare en (paternal eller mors) allel og arbeidet er mer enn tilfredsstillende. Den ikke-aktive allelen kalles stille, nettopp fordi den eksisterer, men den "uttrykker ikke" seg selv.
Cellene i hjernen inneholder alle kromosom 15, men i noen regioner uttrykkes kun den materniske genetiske linjen, mens i andre bare den faderlige. I hypothalamus, som er hjerneområdet som er ansvarlig for Prader-Willi syndrom, uttrykkes bare genene av faderkromosomet normalt.
CHROMOSOMA 15 OG PRADER-WILLI SYNDROME
Gjennom genetisk testing ble det funnet at pasienter med Prader-Willi syndrom mangler et normalt paternal kromosom 15. Dette er skadelig i hypothalamus, hvor den eneste aktive kromosomale linjen er den faderlige.
Hypothalamus hovedfunksjoner:
- Appetittregulering
- Justering av søvnvåkne rytmer
- Uttrykk av følelsesmessige tilstander
- Kroppstemperaturregulering
- Hormonproduksjon
Men hva utløser feilen eller fraværet av faderkromosomen 15? Det er minst tre mulige årsaker:
- Fravær av en bestemt region av faderkromosomen 15: Faktisk mangler kromosomet en vesentlig del.
- To kromosomer 15 mødre. Denne anomali oppstår på grunn av en feil under dannelsen av embryoet.
- Endring av noen gener som er tilstede på paternal kromosom 15.
GENETISK OG HEREDITÆR? ELLER KUN GENETIKK?
Genetikere anser Prader-Willi syndrom en genetisk sykdom, ettersom det fremgår av studier at de tre nevnte mutasjonsmåten ikke arves fra foreldrene (som har en normal kromosom outfit), men oppstår ved en tilfeldighet, like før unnfangelse ( sporadisk mutasjon ).
Imidlertid undergraves sannheten av denne utsagnet ved påvisning av noen par foreldre med mer enn ett barn som lider av Prader-Willi syndrom. I disse tilfellene er det grunn til å tro at det kan være noen arvelig komponent som fremdeles skal demonstreres ved sykdommens opprinnelse.
PRADER-WILLI SYNDROME OG ANGELMAN'S SYNDROME
Prader-Willi syndromet er på noen måter motsatt av Angelman syndrom : i det siste er det faktisk morsmomentet 15 som ikke fungerer som det skal.
Symptomer og komplikasjoner
For å lære mer: Symptomer Prader-Willi syndrom
Prader-Willi syndrom manifesterer seg, med de første symptomene og tegnene, allerede i den tidligste barndommen (første livsår); i denne perioden forårsaker det hovedsakelig en reduksjon i muskeltonen (hypotoni) og en utviklingsforsinkelse. Over tid gjennomgår sykdommen en slags evolusjon, noe som ytterligere beriker det symptomatiske bildet.
OPPVEKST
Hovedtegnene, i løpet av det første år av livet, er:
- Muskelhypotoni . Det betyr at tonen i musklene er redusert i forhold til normal: det er vanligvis avslørt med myke og ikke veldig reaktive lemmer, samt med en vanskelig suging av morsmelk.
- Utviklingsforsinkelse . Det har en tendens til å bli favorisert av sugeproblemer på grunn av hypotoni.
- Strabismus .
- Karakteristiske ansiktsegenskaper . Almond øyne, innsnevring av hodet på templene, munnen nedover og tynn overleppe.
- Delvis eller fullstendig fravær av respons på stimuli . Barnet er slitent og det er ikke lett å vekke ham opp.
Fra alderen til alderen VOKSEN
Siden det første år av livet har det oppstått en lang rekke problemer, noe som kan ha dramatiske resultater.
- Bemerkelsesverdig appetitt og fedme . Pasienter viser et konstant ønske om mat, noe som fører dem til å spise mye og få betydelig vekt. Hvis de ikke finner noe å spise, kommer de til å forbruke frossen mat og avfall, med andre ord, noe spiselig. Alt dette skyldes de forandrede funksjonene til hypothalamus.
- Hypogonadisme . Det betyr at kjønnsorganene (testikler, hos mennesker og eggstokkene hos kvinner) produserer få kjønnshormoner (mannlig testosteron og kvinnelig østrogen). Pasienten fullfører ikke pubertetutvikling og er vanligvis ikke fruktbar. Den første menstruasjonen, hos kvinner, er forsinket (hvis ikke helt fraværende); hos mennesker er det ikke observert noen stemmeforandring.
- Redusert vekst og utvikling . Til problemet med muskelhypotoni, som gjenstår, legges en redusert statural utvikling, selv etter puberteten (hvor vanligvis ungdommer stiger flere centimeter).
- Læreunderskudd . Pasientens intellektuelle evner er nesten alltid redusert.
- Behavioral problemer . Spesielt i ungdomsårene er individer stædige, lunefull og lider av den såkalte obsessive-tvangssykdommen.
- Motorforsinkelse . Barn lærer å gå veldig sent.
- Språkproblemer . Vanligvis begynner pasienten å snakke med betydelig forsinkelse, og deres språk forblir alltid dårlig og vanskelig.
- Søvnforstyrrelser . Den normale vekslingen mellom REM og NON-REM søvnfaser respekteres ikke. Videre, pasienter, når de sover, lider av pusteavbrudd (søvnapné).
- Skoliose . Problemet er kun reservert for enkelte pasienter.
Når du skal henvise til legen
Hos barnet. Tegnene som bør be deg om å mistenke Prader-Willi syndrom er: mangel på utvikling, muskulær hypotoni, problemer med å suge brystmelk, ansiktsegenskaper og mangel på respons på stimuli.
I barnet. To grunnleggende ledetråder er: det vedvarende søket etter mat og den raske vektøkningen.
KOMPLIKASJONER
De viktigste komplikasjonene ved Prader-Willi syndrom skyldes fedme og alle relaterte problemer, som diabetes, hjertesykdom, hypertensjon, hyperkolesterolemi, aterosklerose etc. Videre gjenstår det i sammenheng med kontinuerlig ernæring, det er lett for pasienten å kveles på grunn av et måltid som er forbruket med frodighet.
En annen serie av svært viktige komplikasjoner er relatert til hypogonadisme : mangelen på kjønnshormoner fører ofte til sterilitet og osteoporose .
diagnose
Før man benytter seg av genetiske tester, kan en korrekt pre-diagnose av Prader-Willi syndrom også etableres ved hjelp av en enkel fysisk undersøkelse og noen blodprøver.
Kliniske tegn på fysisk undersøkelse
hos barnet:
- Muskelhypotoni
- Almond øyne
- Krymping av templene
I barnet / ungdommen:
- Uovervinnelig appetitt
- fedme
- Behavioral problemer
Genetiske tester tjener som bekreftelse og bidrar til å avklare hvilken type mutasjon som forårsaket sykdommen.
behandling
Dessverre, siden det er en genetisk sykdom, er Prader-Willi syndrom ikke herdbart.
De eneste anvendelige terapeutiske behandlinger er å begrense symptomer (for eksempel fedme), moderat noen unormal oppførsel og generelt forbedre pasientens levestandard.
For å lykkes i alt dette er det tilrådelig å henvende seg til et team av leger og eksperter, spesialisert på ulike områder, fra endokrinologi til diettologi, fra fysioterapi til psykoterapi.
De vanligste terapeutiske tiltakene er oppført nedenfor.
NÆRING I BARNEN OG I NESTE STEG
Under tidlig spedbarn, for å overvinne vanskeligheter med suging og mangel på utvikling, er det godt å gi barnet høyt kaloriinnhold.
I de følgende faser endres situasjonen helt: maten som administreres må nøye kontrolleres, og maksimal oppmerksomhet til kaloriene.
Den mest egnede spesialisten å spørre om råd er diettisten .
VÆKST HORMONE
Den eksogene administrasjonen (dvs. fra utsiden) av veksthormonet ( GH ) har tre virkninger:
- Oppmuntre til vekst, noe som ellers ville mangle
- Forbedre muskeltonen
- Reduser kroppsfettnivået
Behandlingen starter i en alder av 3-5 år.
I dag er det hormonelle preparater opprettet i laboratoriet, effektive og med reduserte bivirkninger.
Den mest indikerte spesialisten, i dette tilfellet, er en endokrinolog .
SEXUELLE HORMONER
Den eksogene administrasjonen av testosteron, for menn og østrogener, for kvinner, er viktig for å gjenopprette de reduserte nivåene av disse to hormonene. I tillegg til å forbedre fruktbarheten, har hormonbehandling også effekter mot osteoporose.
Behandlingen starter ved puberteten.
For å lære mer: Narkotika for behandling av Prader-Willi syndrom »
FYSIOTERAPI OG LOGOPEDIA
Pasienter med Prader-Willi syndrom trenger fysisk og språklig rehabilitering . Den første tar sikte på å begrense muskulær hypotoni og effekten av fedme; Det andre middelet til kommunikasjonsmangler, både muntlig og skriftlig.
Eksperter, som skal vende seg til, er henholdsvis en fysioterapeut og en taleterapeut.
Sykdoterapi og sysselsettingsterapi
Psykoterapi er viktig for de pasientene med obsessiv-kompulsive lidelser og humør generelt. Støtten til en psykiater eller en psykolog kan i stor grad forbedre adferdsaspektet.
Arbeidsterapi, derimot, tar sikte på å lære pasienten hvordan man skal ta vare på seg selv, hvordan å kle seg etc., med andre ord hvordan man skal utføre de viktigste daglige aktivitetene.
HJELPEN AV FAMILIER
Nærheten til familiemedlemmer er viktig for å hjelpe de syke slektninger, særlig under ungdommen. Rådene som vanligvis er gitt til familier, er å følge pasienten i alle sine aktiviteter (spesielt når han er lei), for å spørre om den mest hensiktsmessige oppførselen for å reservere for ham, ikke å ekskludere ham osv.
Prognose og forebygging
Gitt at Prader-Willi syndrom er en uhelbredelig sykdom, kan prognosen aldri være positiv. Den største faren er representert av fedme og av de patologiene som er forbundet med det: døden skyldes vanligvis en av disse.
De tilgjengelige behandlingene (balansert kosthold, hormonbehandling, psykoterapi, etc.) forbedrer livskvaliteten, selv på en sensitiv måte. Det er imidlertid fortsatt tiltak for symptomkontroll og ingenting mer.
Nærheten til slektninger er grunnleggende: deres støtte kan faktisk forlenge pasientens liv.
FOREBYGGING
Når sykdommen oppstår i embryoet på grunn av en genetisk mutasjon, er det ingen måte å forhindre det på.
Hvis to foreldre i stedet har født et barn med Prader-Willi syndrom, kan de under en graviditet gjennomgå spesifikke genetiske tester for å finne ut om de er bærere av sykdommen eller ikke.


